Plant Biology Highlights: Nature Articles 2017
It's almost the end of another amazing year. Undoubtedly, we came across amazing plant science stories all the year round. Like all other researchers, I regularly follow plant-specific journals (The Plant Cell, Plant Physiology, Nature Plants, The Plant Journal, Journal of Experimental Botany, Molecular Plant, Plant, Cell & Environment, Plant, Cell & Physiology, Frontiers in Plant Science, Plant Direct and so on). Apart from that Cell, Science, Nature, PNAS, Nature Communication and other renowned journals cover plant science stories. At the end of this 2017, I've covered few great stories from Nature in this post.
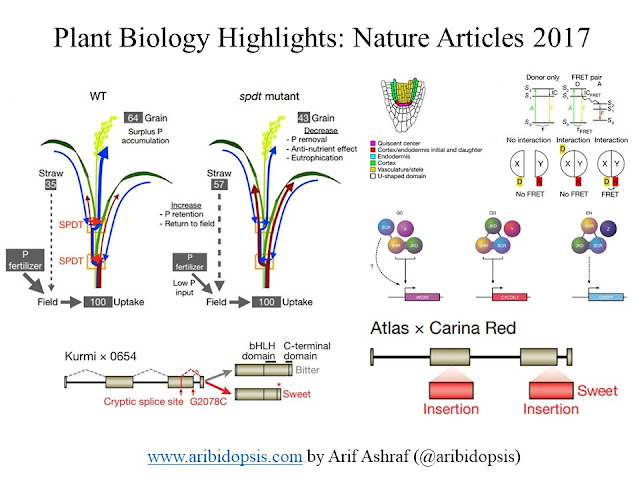
Reducing phosphorus accumulation in rice grains with an impaired transporter in the node

Genome sequence and genetic diversity of European ash trees
Ash trees are really popular for building materials such as baseball bats, bodies of guitars. It's very useful to lit fires and keeps the fire for a longer time, for instances barbeque. Unfortunately, this such a useful tree is under prominent threads. Millions of ash trees had been destroyed due to the fungus (Hymenoscyphus fraxineus) in Europe and the herbivorous beetle (Agrilus planipennis) in North America.

In this paper, they published the genome sequence of Ash tree (Fraxinus excelsior) from the UK and pertaining analysis. At the same time, they sequenced ash trees from the region where it is susceptible to ash dieback (ADB), caused by fungus Hymenoscyphus fraxineus, and developed a set of markers to identify the ADB susceptibility. This marker set suggests that British populations are less susceptibility to ADB compared to Denmark. They have also identified the chemical, Iridoid glycoside, as a classifier to distinguish between ADB susceptible and non-susceptible populations.
The genome of Chenopodium quinoa
We are facing the food crisis for the growing population in many parts of the globe. To circumvent the situation, we often talk about better yielding, biotic and abiotic stress tolerant crops and alternative source of food. Chenopodium quinoa is one of the potential sources as an alternative food. Its seed is edible and contains a balanced amount of essential amino acids, fiber, lipids, carbohydrates, vitamins, and minerals. Additionally, it is gluten-free and has a low glycaemic index. This excellent choice of alternative food source was got attention due to the declaration of the International Year of Quinoa by United Nations in 2013. In this paper, they have published the genome of Chenopodium quinoa and unravel the genetic explanation behind sweet varieties.

They have compared bitter (Kurmi×0654) and sweet (Atlas×Carina Red) varieties to find the genetic circuit. They have found the triterpene saponin biosynthesis activating regulator 1 (TSAR1), a bHLH transcription factor, as a pivotal point. A premature stop codon gives the rise of the sweet variety. The comprehensive knowledge from the genome sequence will help to achieve desired traits such as shorter plants, fewer branches, compact seed heads, increased heat and biotic stress tolerance, and the introgression of the sweet phenotype into commercial varieties.
Root microbiota drive direct integration of phosphate immunity and stress
Plants reside with soil microbes and compete for nutrients as well as. In this paper, they used limited phosphate condition to induce phosphate starvation responses (PSR) and synthetic bacterial community (SynCom) to simulate the bacterial presence to compete for the phosphate. When they grew plants under limited phosphate condition, they have found that SynCom induces the activity of transcriptional regulator PHOSPHATE STARVATION RESPONSE 1 (PHR1). Interestingly, PHR1 is the major regulator of PSR. The major finding of this work is to pinpoint PHR1 as a convergence point between phosphate starvation and immune response.
For further details, you may visit the website of Dangl Lab.
A chromosome conformation capture ordered sequence of the barley genome
Barley is one of the major crops which is important also from agricultural revolution perspective as several archaeological sites revealed that fact in recent years. Additionally, it may have a connection to inspire the first brewing of beer. This paper published a highly contiguous reference genome sequence for barley by combining hierarchical shotgun sequencing with novel technologies such as optical mapping and chromosome-scale scaffolding with Hi-C.
In this study, they showed that barley contains 23 members of SUGARS WILL EVENTUALLY BE EXPORTED TRANSPORTER (SWEET) transmembrane proteins. few of them (SWEET11, SWEET13, SWEET14, and SWEET15) have two or more genes for each subgroup compared with only a single orthologue in rice and Arabidopsis. For instances, both SWEET11a and SWEET11b were highly expressed in maternal seed tissue. This example of SWEET gene highlights the importance of the high-quality reference genome sequence to unravel the evolutionary history of gene duplications, their relation to morphological and physiological innovations, and their impact on crop performance.
The sunflower genome provides insights into oil metabolism, flowering, and Asterid evolution
Sunflower is the global source of oil. Apart from that, it has better adaptation capability in such a detrimental global climate. In this paper, they have published the genome sequence of domesticated sunflower Helianthus annuus L. They took an integrative approach combining quantitative genetics, expression and diversity data to decipher the gene networks for two major breeding traits: flowering time and oil metabolism.
For the flowering time, they compared it with the recently developed database of flowering-time gene networks in Arabidopsis thaliana. They identified 485 orthologues and in-paralogues for 270 flowering-time genes in the sunflower genome. To decipher the genetic bases of oil metabolism, they reconstructed a genome-scale metabolic network for the sunflower and extracted metabolic pathways involved in oil synthesis, yielding a total of 429 genes mapped onto 125 reactions, corresponding to 12 pathways. From there, they narrowed down to 46 oil genes in 32 genomic regions corresponding to previously identified QTLs for seven oil-related traits. Nine of these genes were highly differentiated between high and low-oil lines, including FAD2-1, which has been shown to be under selection during post-domestication. HPPD had already been found to co-localize with a QTL for the vitamin E precursor tocopherol.
Improved maize reference genome with single-molecule technologies
Complete and accurate reference genomes and annotations are the rudimentary resources for the determination of biological processes and support translation of research findings into improved and sustainable agricultural technologies. In this article they reported the assembly and annotation of a reference genome of maize, a genetic and agricultural model species, using single-molecule real-time sequencing and high-resolution optical mapping. Compared to the previous reference genome, this assembly features a 52-fold increase in contig length and notable improvements in the assembly of intergenic spaces and centromeres. Characterization of the repetitive portion of the genome revealed more than 130,000 intact transposable elements, allowing them to identify transposable element lineage expansions that are unique to maize. Gene annotations were updated using 111,000 full-length transcripts obtained by single-molecule real-time sequencing. In addition, comparative optical mapping of two other inbred maize lines revealed a prevalence of deletions in regions of low gene density and maize lineage-specific genes
In vivo FRET-FLIM reveals cell-type-specific protein interactions in Arabidopsis root
To understand the primary root growth development of Arabidopsis thaliana, we need temporal and spatial data of gene expression and protein-protein interactions. Elavorately, the protein-protein interactions, and gene expression hold the mechanistic explanation for the determination of cell fate. For example, SHORT-ROOT (SHR), SCARECROW (SCR), and JACKDAW (JKD) - these three transcription factors co-expressed and interacts as well as. SHR transcription factor moves to the U-shaped domain from the vasculature and induces expression of SCR to promote divisions that separate cortex and endodermis by activating CYCLIN D6;1 (CYCD6;1) in the CEI (Cortex/Endodermis Initials).

They have selected three genes specific for three cell types: QC (WOX5), CEI (CYCD6;1), and Endodermis (CASP1). And, they wanted to investigate how the interaction of this three transcription factor regulates cell fate. The above figure shows the spatial protein-protein interactions-based cell fate regulation. Overall, this in vivo FRET–FLIM analysis reveals that SHR, SCR, and JKD form qualitatively different higher-order transcription factor complexes. These distinct complexes associate with target promoter selectivity and cell-fate segregation between closely neighboring cells.
GLUTAMATE RECEPTOR-LIKE channels are essential for chemotaxis and reproduction in mosses
Glutamate receptors are well characterized channels that mediate cell-to-cell communication during neurotransmission in animals, but their functional role in organisms without a nervous system remains unclear. In plants, genes of the GLUTAMATE RECEPTOR-LIKE (GLR) family have been implicated in defence against pathogens, reproduction, control of stomata aperture and light signal transduction. However, the large number of GLR genes present in angiosperm genomes (20 to 70) has prevented the observation of strong phenotypes in loss-of-function mutants. Here they showed that in the basal land plant Physcomitrella patens, mutation of the GLR genes GLR1 and GLR2 causes failure of sperm cells to target the female reproductive organs. In addition, they observed that GLR genes encode non-selective Ca2+-permeable channels that can regulate cytoplasmic Ca2+ and are needed to induce the expression of a BELL1-like transcription factor essential for zygote development. This work reveals functions for GLR channels in sperm chemotaxis and transcriptional regulation. Sperm chemotaxis is essential for fertilization in both animals and early land plants such as bryophytes and pteridophytes. Therefore, these results suggest that ionotropic glutamate receptors may have been conserved throughout plant evolution to mediate cell-to-cell communication during sexual reproduction.
The Apostasia genome and the evolution of orchids
Constituting approximately 10% of flowering plant species, orchids (Orchidaceae) display unique flower morphologies, possess an extraordinary diversity in lifestyle, and have successfully colonized almost every habitat on Earth. Here they reported the draft genome sequence of Apostasia shenzhenica, a representative of one of two genera that form a sister lineage to the rest of the Orchidaceae, providing a reference for inferring the genome content and structure of the most recent common ancestor of all extant orchids and improving our understanding of their origins and evolution. In addition, they presented transcriptome data for representatives of Vanilloideae, Cypripedioideae and Orchidoideae, and novel thirdgeneration genome data for two species of Epidendroideae, covering all five orchid subfamilies. A. shenzhenica shows clear evidence of a whole-genome duplication, which is shared by all orchids and occurred shortly before their divergence. Comparisons between A. shenzhenica and other orchids and angiosperms also permitted the reconstruction of an ancestral orchid gene toolkit. They identified new gene families, gene family expansions and contractions, and changes within MADS-box gene classes, which control a diverse suite of developmental processes, during orchid evolution. This study sheds new light on the genetic mechanisms underpinning key orchid innovations, including the development of the labellum and gynostemium, pollinia, and seeds without endosperm, as well as the evolution of epiphytism; reveals relationships between the Orchidaceae subfamilies, and helps clarify the evolutionary history of orchids within the angiosperms.
Genome sequence of the progenitor of the wheat D genome Aegilops tauschii
Aegilops tauschii is the diploid progenitor of the D genome of hexaploid wheat (Triticum aestivum, genomes AABBDD) and an important genetic resource for wheat. The large size and highly repetitive nature of the Ae. tauschii genome has until now precluded the development of a reference-quality genome sequence. Here, they used an array of advanced technologies, including ordered-clone genome sequencing, whole-genome shotgun sequencing, and BioNano optical genome mapping, to generate a reference quality genome sequence for Ae. tauschii ssp. strangulata accession AL8/78, which is closely related to the wheat D genome. They showed that compared to other sequenced plant genomes, including a much larger conifer genome, the Ae. tauschii genome contains unprecedented amounts of very similar repeated sequences. This genome comparisons reveal that the Ae. tauschii genome has a greater number of dispersed duplicated genes than other sequenced genomes and its chromosomes have been structurally evolving an order of magnitude faster than those of other grass genomes. The decay of colinearity with other grass genomes correlates with recombination rates along chromosomes. They proposed that the vast amounts of very similar repeated sequences cause frequent errors in recombination and lead to gene duplications and structural chromosome changes that drive fast genome evolution.
For more exciting plant biology stories, you may follow Nature Plants. If you are interested to take a look of plant biology stories from Nature in 2016, please go through the following post:
Plant Biology Highlights: Nature Articles 2016

Reducing phosphorus accumulation in rice grains with an impaired transporter in the node
Phosphorus is one of the most indispensable minerals required by plants and animals. In the human body, the strength of bones and teeth is provided by mostly calcium and phosphorus. Plants, including crop plants like rice, have transporters to uptake phosphorus. And, the majority portion (~60%) of phosphorus in rice finds their way towards the grains, which is consumed by humans and poultry. Unfortunately, we are unable to digest phytate, one of the most abundant forms of phosphorus in grains, and excrete to lead their way to flow through water eventually. As a result, water has an overload of nutrients, popularly termed as Eutrophication, and due to excessive growth of algae and other stuff, there is depletion of oxygen.

Jian Feng Ma and his team in the Okayama University, Japan found a transporter, SULTR-like Phosphorus Distribution Transporter (SPDT), in rice which controls phosphorus accumulation in the grain. Knock out of this particular gene reduces phosphorus accumulation in grain and at the same time, keep other parameters related to growth and development unaltered. manipulation of this transporter shows the promise to make phosphorus balance.
Genome sequence and genetic diversity of European ash trees
Ash trees are really popular for building materials such as baseball bats, bodies of guitars. It's very useful to lit fires and keeps the fire for a longer time, for instances barbeque. Unfortunately, this such a useful tree is under prominent threads. Millions of ash trees had been destroyed due to the fungus (Hymenoscyphus fraxineus) in Europe and the herbivorous beetle (Agrilus planipennis) in North America.

In this paper, they published the genome sequence of Ash tree (Fraxinus excelsior) from the UK and pertaining analysis. At the same time, they sequenced ash trees from the region where it is susceptible to ash dieback (ADB), caused by fungus Hymenoscyphus fraxineus, and developed a set of markers to identify the ADB susceptibility. This marker set suggests that British populations are less susceptibility to ADB compared to Denmark. They have also identified the chemical, Iridoid glycoside, as a classifier to distinguish between ADB susceptible and non-susceptible populations.
The genome of Chenopodium quinoa
We are facing the food crisis for the growing population in many parts of the globe. To circumvent the situation, we often talk about better yielding, biotic and abiotic stress tolerant crops and alternative source of food. Chenopodium quinoa is one of the potential sources as an alternative food. Its seed is edible and contains a balanced amount of essential amino acids, fiber, lipids, carbohydrates, vitamins, and minerals. Additionally, it is gluten-free and has a low glycaemic index. This excellent choice of alternative food source was got attention due to the declaration of the International Year of Quinoa by United Nations in 2013. In this paper, they have published the genome of Chenopodium quinoa and unravel the genetic explanation behind sweet varieties.

They have compared bitter (Kurmi×0654) and sweet (Atlas×Carina Red) varieties to find the genetic circuit. They have found the triterpene saponin biosynthesis activating regulator 1 (TSAR1), a bHLH transcription factor, as a pivotal point. A premature stop codon gives the rise of the sweet variety. The comprehensive knowledge from the genome sequence will help to achieve desired traits such as shorter plants, fewer branches, compact seed heads, increased heat and biotic stress tolerance, and the introgression of the sweet phenotype into commercial varieties.
Root microbiota drive direct integration of phosphate immunity and stress
Plants reside with soil microbes and compete for nutrients as well as. In this paper, they used limited phosphate condition to induce phosphate starvation responses (PSR) and synthetic bacterial community (SynCom) to simulate the bacterial presence to compete for the phosphate. When they grew plants under limited phosphate condition, they have found that SynCom induces the activity of transcriptional regulator PHOSPHATE STARVATION RESPONSE 1 (PHR1). Interestingly, PHR1 is the major regulator of PSR. The major finding of this work is to pinpoint PHR1 as a convergence point between phosphate starvation and immune response.
For further details, you may visit the website of Dangl Lab.
A chromosome conformation capture ordered sequence of the barley genome
Barley is one of the major crops which is important also from agricultural revolution perspective as several archaeological sites revealed that fact in recent years. Additionally, it may have a connection to inspire the first brewing of beer. This paper published a highly contiguous reference genome sequence for barley by combining hierarchical shotgun sequencing with novel technologies such as optical mapping and chromosome-scale scaffolding with Hi-C.
In this study, they showed that barley contains 23 members of SUGARS WILL EVENTUALLY BE EXPORTED TRANSPORTER (SWEET) transmembrane proteins. few of them (SWEET11, SWEET13, SWEET14, and SWEET15) have two or more genes for each subgroup compared with only a single orthologue in rice and Arabidopsis. For instances, both SWEET11a and SWEET11b were highly expressed in maternal seed tissue. This example of SWEET gene highlights the importance of the high-quality reference genome sequence to unravel the evolutionary history of gene duplications, their relation to morphological and physiological innovations, and their impact on crop performance.
The sunflower genome provides insights into oil metabolism, flowering, and Asterid evolution
Sunflower is the global source of oil. Apart from that, it has better adaptation capability in such a detrimental global climate. In this paper, they have published the genome sequence of domesticated sunflower Helianthus annuus L. They took an integrative approach combining quantitative genetics, expression and diversity data to decipher the gene networks for two major breeding traits: flowering time and oil metabolism.
For the flowering time, they compared it with the recently developed database of flowering-time gene networks in Arabidopsis thaliana. They identified 485 orthologues and in-paralogues for 270 flowering-time genes in the sunflower genome. To decipher the genetic bases of oil metabolism, they reconstructed a genome-scale metabolic network for the sunflower and extracted metabolic pathways involved in oil synthesis, yielding a total of 429 genes mapped onto 125 reactions, corresponding to 12 pathways. From there, they narrowed down to 46 oil genes in 32 genomic regions corresponding to previously identified QTLs for seven oil-related traits. Nine of these genes were highly differentiated between high and low-oil lines, including FAD2-1, which has been shown to be under selection during post-domestication. HPPD had already been found to co-localize with a QTL for the vitamin E precursor tocopherol.
Improved maize reference genome with single-molecule technologies
Complete and accurate reference genomes and annotations are the rudimentary resources for the determination of biological processes and support translation of research findings into improved and sustainable agricultural technologies. In this article they reported the assembly and annotation of a reference genome of maize, a genetic and agricultural model species, using single-molecule real-time sequencing and high-resolution optical mapping. Compared to the previous reference genome, this assembly features a 52-fold increase in contig length and notable improvements in the assembly of intergenic spaces and centromeres. Characterization of the repetitive portion of the genome revealed more than 130,000 intact transposable elements, allowing them to identify transposable element lineage expansions that are unique to maize. Gene annotations were updated using 111,000 full-length transcripts obtained by single-molecule real-time sequencing. In addition, comparative optical mapping of two other inbred maize lines revealed a prevalence of deletions in regions of low gene density and maize lineage-specific genes
In vivo FRET-FLIM reveals cell-type-specific protein interactions in Arabidopsis root
To understand the primary root growth development of Arabidopsis thaliana, we need temporal and spatial data of gene expression and protein-protein interactions. Elavorately, the protein-protein interactions, and gene expression hold the mechanistic explanation for the determination of cell fate. For example, SHORT-ROOT (SHR), SCARECROW (SCR), and JACKDAW (JKD) - these three transcription factors co-expressed and interacts as well as. SHR transcription factor moves to the U-shaped domain from the vasculature and induces expression of SCR to promote divisions that separate cortex and endodermis by activating CYCLIN D6;1 (CYCD6;1) in the CEI (Cortex/Endodermis Initials).

They have selected three genes specific for three cell types: QC (WOX5), CEI (CYCD6;1), and Endodermis (CASP1). And, they wanted to investigate how the interaction of this three transcription factor regulates cell fate. The above figure shows the spatial protein-protein interactions-based cell fate regulation. Overall, this in vivo FRET–FLIM analysis reveals that SHR, SCR, and JKD form qualitatively different higher-order transcription factor complexes. These distinct complexes associate with target promoter selectivity and cell-fate segregation between closely neighboring cells.
GLUTAMATE RECEPTOR-LIKE channels are essential for chemotaxis and reproduction in mosses
Glutamate receptors are well characterized channels that mediate cell-to-cell communication during neurotransmission in animals, but their functional role in organisms without a nervous system remains unclear. In plants, genes of the GLUTAMATE RECEPTOR-LIKE (GLR) family have been implicated in defence against pathogens, reproduction, control of stomata aperture and light signal transduction. However, the large number of GLR genes present in angiosperm genomes (20 to 70) has prevented the observation of strong phenotypes in loss-of-function mutants. Here they showed that in the basal land plant Physcomitrella patens, mutation of the GLR genes GLR1 and GLR2 causes failure of sperm cells to target the female reproductive organs. In addition, they observed that GLR genes encode non-selective Ca2+-permeable channels that can regulate cytoplasmic Ca2+ and are needed to induce the expression of a BELL1-like transcription factor essential for zygote development. This work reveals functions for GLR channels in sperm chemotaxis and transcriptional regulation. Sperm chemotaxis is essential for fertilization in both animals and early land plants such as bryophytes and pteridophytes. Therefore, these results suggest that ionotropic glutamate receptors may have been conserved throughout plant evolution to mediate cell-to-cell communication during sexual reproduction.
The Apostasia genome and the evolution of orchids
Constituting approximately 10% of flowering plant species, orchids (Orchidaceae) display unique flower morphologies, possess an extraordinary diversity in lifestyle, and have successfully colonized almost every habitat on Earth. Here they reported the draft genome sequence of Apostasia shenzhenica, a representative of one of two genera that form a sister lineage to the rest of the Orchidaceae, providing a reference for inferring the genome content and structure of the most recent common ancestor of all extant orchids and improving our understanding of their origins and evolution. In addition, they presented transcriptome data for representatives of Vanilloideae, Cypripedioideae and Orchidoideae, and novel thirdgeneration genome data for two species of Epidendroideae, covering all five orchid subfamilies. A. shenzhenica shows clear evidence of a whole-genome duplication, which is shared by all orchids and occurred shortly before their divergence. Comparisons between A. shenzhenica and other orchids and angiosperms also permitted the reconstruction of an ancestral orchid gene toolkit. They identified new gene families, gene family expansions and contractions, and changes within MADS-box gene classes, which control a diverse suite of developmental processes, during orchid evolution. This study sheds new light on the genetic mechanisms underpinning key orchid innovations, including the development of the labellum and gynostemium, pollinia, and seeds without endosperm, as well as the evolution of epiphytism; reveals relationships between the Orchidaceae subfamilies, and helps clarify the evolutionary history of orchids within the angiosperms.
Genome sequence of the progenitor of the wheat D genome Aegilops tauschii
Aegilops tauschii is the diploid progenitor of the D genome of hexaploid wheat (Triticum aestivum, genomes AABBDD) and an important genetic resource for wheat. The large size and highly repetitive nature of the Ae. tauschii genome has until now precluded the development of a reference-quality genome sequence. Here, they used an array of advanced technologies, including ordered-clone genome sequencing, whole-genome shotgun sequencing, and BioNano optical genome mapping, to generate a reference quality genome sequence for Ae. tauschii ssp. strangulata accession AL8/78, which is closely related to the wheat D genome. They showed that compared to other sequenced plant genomes, including a much larger conifer genome, the Ae. tauschii genome contains unprecedented amounts of very similar repeated sequences. This genome comparisons reveal that the Ae. tauschii genome has a greater number of dispersed duplicated genes than other sequenced genomes and its chromosomes have been structurally evolving an order of magnitude faster than those of other grass genomes. The decay of colinearity with other grass genomes correlates with recombination rates along chromosomes. They proposed that the vast amounts of very similar repeated sequences cause frequent errors in recombination and lead to gene duplications and structural chromosome changes that drive fast genome evolution.
For more exciting plant biology stories, you may follow Nature Plants. If you are interested to take a look of plant biology stories from Nature in 2016, please go through the following post:
Plant Biology Highlights: Nature Articles 2016





Comments
Post a Comment